
Hendrik Dietz: ATP synthase is a nanofactory. Look at that enzyme. As enzymes go, it’s huge. The vast number of ATP synthase molecules in a human body make 50 lbs of ATP every day. They are in the middle of the gap in the humans’ ability to engineer matter: At 20 nm, they are too small for photolithography (computer chip manufacturing). Yet they are also too big for synthetic chemistry. DNA origami is about the only tool we have to precisely engineer things at this scale. The detailed principles of protein folding, even of 50 year old protein structures, are still unknown. Could we be close with DNA origami to making functional structures?
The Dietz lab made a completely asymmetric structure at about 20 nm. TEM tomography shows its shape is accurate to the design. It is made of about 440,000 atoms. For a molecule, it is very, very big. Prof. Dietz had a 3D printed model to compare to a 3D printed ribosome. The designed molecule is significantly bigger.
The number of unpaired bases in a structure designed with fully paired DNA is a good metric of the quality of the folding. Their Assay says that different origami structures with full complement of staples have 40-260 unpaired bases. Looking at the image, the defects are not visible. That also means that defects are hidden in every origami picture ever. Several stapples are missing, on average, but the structures still fold.
I was very interested in their 2 applications:
1: These big origami structures can act as an alignment jig for molecules to be examined by electron microscopy. Normally smallish proteins are too small even for electron microscopy, but if you frame them and constrain their motion, you can get EM data. If you average it, you can get a structure. The resolution is not great, but the fact that is can be done is amazing.
2: These molecules can enhance 2-beam optical trapping analysis of molecular unfolding. They use rigid origami rods instead of floppy single-stranded DNA. This can directly measure the tiny energies associated with folding and unfolding (~ 1.4 kbT). The way it was presented was quite intuitive.
Alireza Goudarzi (and Darko Stefanovic): A group of reactions can act as a neural network. Normally, in a neural net, you can adjust the parameters inside the network to get the whole network to “learn.” I don’t really understand “training” in this system. I gather that the chaotic system will tend to sync to inputs. Maybe it is similar to “Feature extraction” in machine learning?
Ralf Jungmann: Light is limited to imaging things larger than its own wavelength (broadly speaking). For smaller objects (below 200-400 nm) we need to use things like an electron microscope (samples that are frozen or in vacuum or both). Recently, people have engineered other superresolution tricks that get light to break its normal rules. The trick presented (DNA PAINT) uses a molecular beacon that can pop on and off of a set of immobile binding sites. Since there are so few molecules, you can fit the center of the Gaussian for each one individually. Places where the centers of individual Gaussians align are the immobile binding sites. This technique looks extremely pretty. A 5 min movie of transient DNA binding and unbinding (DNA PAINT) allows localization of binding sites to a few nanometers.
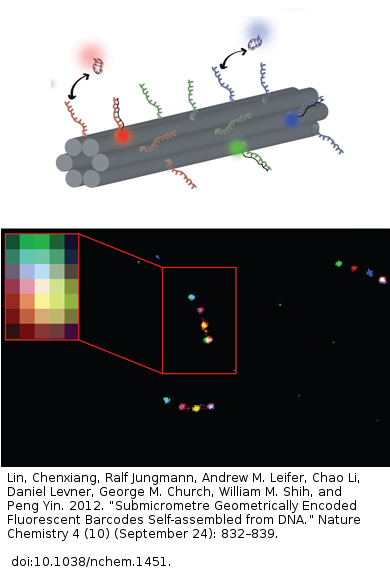
Application as geometric barcoding: DNA PAINT allows 215 different barcoded ~10 nm structures that can be identified by PAINT. They are still not much bigger than antibodies. The subject must hold still for this to work, so it only will work for fixed cells. But it is seriously impressive. That fuzzy colored thing in the upper left is the normal fluorescence microscopy. That’s roughly what it looks like by eye. Then if you enhance with this technique, you see four different-colored spots along a chain. See dna-paint.org
Carl Brown showed results of integrating DNAzymes into signal cascades. A topologically constrained latent activator is a substrate for a DNAzyme. The DNAzyme displaces the a blocker and then cleaves the latent activator. Once cleaved, the blocker is gone. So it’s now active. Leakage is bad. It cascades like Xi Chen’s work, but it can go to 4 layers due to the DNAzyme being a more specific, less leakage prone system compared to CHA. They are also exploring mismatches to speed up the system in a manner similar to Sherry Jiang here in the Ellington Lab. So that’s interesting.
Chengde Mao compared two approaches to building self assembled objects: 1.) Flexible subunits with connectivity precisely defined; or 2.) more rigid objects with redundant connectivity. I lost the thread as I was working on my presentation.
Yonggang Ke is building structures by adding DNA bricks. The bricks have the same geometry, but different linkage specificity. They seem convenient as one does not need to design staples or geometry or constraints of the scaffold. You just add the different combinations of bricks and then anneal. How many different bricks do they have? I imagine it’s a lot in order to get good yield of the correct structure.
John Sadowski presented tricks to control assembly-order of classic DNA tetrahedrons. With strand-displacement reactions, assembly occurs in sequence. This helps answer the question of “Is everything in your structure where and what you think it is?”
The bricks system presented by Yonggang Ke might be missing whole blocks and it would be impossible to tell on cryo-EM. Dietz showed that definitively. So how can you design an assembly process to only proceed according to a particular sequence of events?
John Sadowski’s system was inspired by embryonic development: it has spatial and temporal control over the assembly process at one temperature with useful responses to the environment. He calls this “developmental self assembly.” He has a lot of really good controls. He definitely answered the question of “is everything there, and is it where you think it is?”
