More science for my students. Yesterday, I showed a fluorescence polarization (FP) binding assay with frustratingly large error bars. The standard deviation among three replicates was disturbingly large. Put another way: three samples that were supposed to be the same looked different from one another. It would be like pouring three glasses of wine from the same bottle only to discover that one glass was red, one white, and one rose.
One possibility is that the samples are actually not as similar as we thought. Just because the three glasses of wine came from the same bottle doesn’t mean they were poured the same way. Maybe the bottle had sediment that made one glass look darker.
The other possibility is that the instrument is just not very good. In my wine analogy, maybe it’s not that the three glasses are different; maybe I just need to get my eyes checked. Or to stop drinking.
I tested that yesterday. The FP assay, not stopping drinking. Heaven forbid.
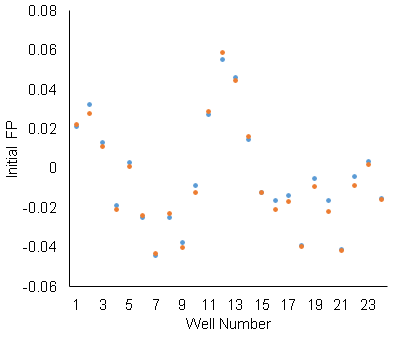
I filled seven wells with the exact same sample. I used a microscope to confirm that there were no bubbles (bubbles play mad hob with the FP measurement). I ran the same plate with 7 identical samples through the machine 15 times. Here’s what it looked like:

What it should look like is seven overlapping flat lines. That word “should” is a dead giveaway that something is amiss. What I see here is a whole lot of noise. How can I correct for this? (short of buying a new plate reader – which I covet)
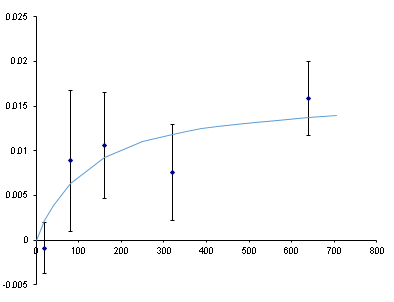
I can compensate by averaging. When I average across all 15 replications, I get one halfway decent measurement. I added protein to the experimental wells and took 15 more measurements. The control sample changed dramatically (-8.5 mP). To be clear, that control sample was not touched between the initial and final measurements. That apparent change in the first sample was introduced by the instrument.

I can use the control as an internal standard to help correct that kind of variability. I took every individual fluorescence measurement and normalized it to the control sample. The run-to-run standard deviation increased from 6mP to 15 mP (presumably because I was adding independent noise from the standard well to the noise in the experimental wells). So there is no systematic across-the-row error. But it does correct the drifting background.
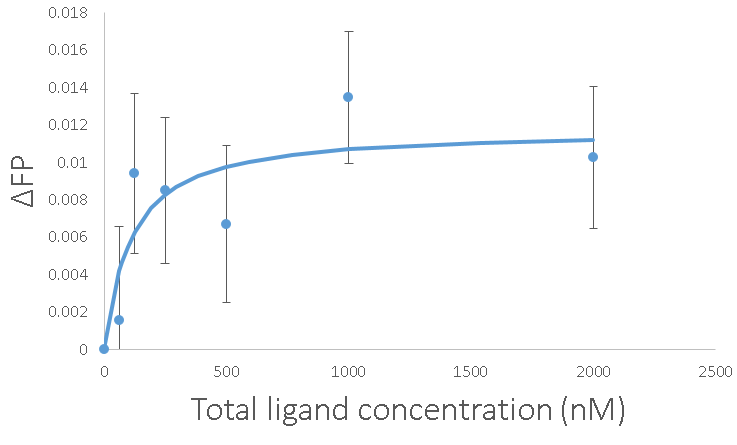
With all of that work, we can get marginally reliable measurements. The error bars are the standard error of the average with n=15. The Kd came out to 100 nM again, which gives me more confidence. Going about it this way has the advantage of being able to get those error bars down just by adding more replicates. It also has the advantage of not costing $20,000 for a new platereader.




You must be logged in to post a comment.